This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
Introduction
Myotonic dystrophy type 2 (DM2) is a form of muscular dystrophy characterized by muscle loss and muscle weakness, along with a wide range of other symptoms including myotonia, early-onset cataracts, heart conduction problems and muscle pain. Myotonic dystrophy is the most common MD in adults, and it affects approximately 1 in every 8000 individuals. The disease follows an autosomal dominant fashion, and symptoms usually do not appear until the individuals 20s or 30s [1].
DM2 is caused by a mutation in the CNBP gene which is located on the long arm of chromosome 3. The specific mutation that causes DM2 is an excess of CCTG nucleotide repeats found within the first intron of the CNBP gene. These repeats number anywhere from 75-11,000 in total for diseased individuals, whereas healthy individuals usually have fewer than 26 repeats in total. These excessive repeats result in the CNBP-pre-mRNA forming a doubled-stranded structure, therefore down regulating the CNBP protein in the disease state [1]. The CNBP protein is highly conserved across muscular organisms, contains seven zinc-finger domains in humans and has been linked to the regulation of transcription and translation. It is found within the cytosol, nucleus and ER [2].
One primary mechanism that contributes to DM2 is an RNA-gain-of-function mechanism from the mutant the CNBP-pre-mRNA. The excessive CCTG repeats in diseased individuals results in the formation of a doubled-stranded RNA structure that sequester pproteins within the cell, thus leading to dysregulation of various processes. In addition, the mutant the CNBP-pre-mRNA is not translated due to its double stranded nature, so CNBP protein levels are also highly down-regulated in the disease state. While is known that mutant CNBP-pre-mRNA can have a wide range of negative effects, it is unclear how decreased CNBP protein levels contributes to muscle wasting and weakness [1]. Therefore, the goal of this study is to look at how low levels of CNBP contributes to muscle wasting and weakness using a mice model.
DM2 is caused by a mutation in the CNBP gene which is located on the long arm of chromosome 3. The specific mutation that causes DM2 is an excess of CCTG nucleotide repeats found within the first intron of the CNBP gene. These repeats number anywhere from 75-11,000 in total for diseased individuals, whereas healthy individuals usually have fewer than 26 repeats in total. These excessive repeats result in the CNBP-pre-mRNA forming a doubled-stranded structure, therefore down regulating the CNBP protein in the disease state [1]. The CNBP protein is highly conserved across muscular organisms, contains seven zinc-finger domains in humans and has been linked to the regulation of transcription and translation. It is found within the cytosol, nucleus and ER [2].
One primary mechanism that contributes to DM2 is an RNA-gain-of-function mechanism from the mutant the CNBP-pre-mRNA. The excessive CCTG repeats in diseased individuals results in the formation of a doubled-stranded RNA structure that sequester pproteins within the cell, thus leading to dysregulation of various processes. In addition, the mutant the CNBP-pre-mRNA is not translated due to its double stranded nature, so CNBP protein levels are also highly down-regulated in the disease state. While is known that mutant CNBP-pre-mRNA can have a wide range of negative effects, it is unclear how decreased CNBP protein levels contributes to muscle wasting and weakness [1]. Therefore, the goal of this study is to look at how low levels of CNBP contributes to muscle wasting and weakness using a mice model.
Aim 1
The purpose of my first aim is to identify the conserved sites of CNBP that are critical to proper muscle function. To do this, I will search for CNBP homologs in various species to identify conserved protein domains using the SMART database. Then, using Mus musculus as a model organism, I will individually make targeted amino acid substitutions using CRISPR/Cas9 within amino acids that are conserved only within muscular organisms near these domains. Following this, I will screen for mice that exhibit muscle wasting and weakness DM2 phenotypes to determine which conserved sites of CNBP are most critical to the proper functioning of skeletal muscle tissue (Figure 1).
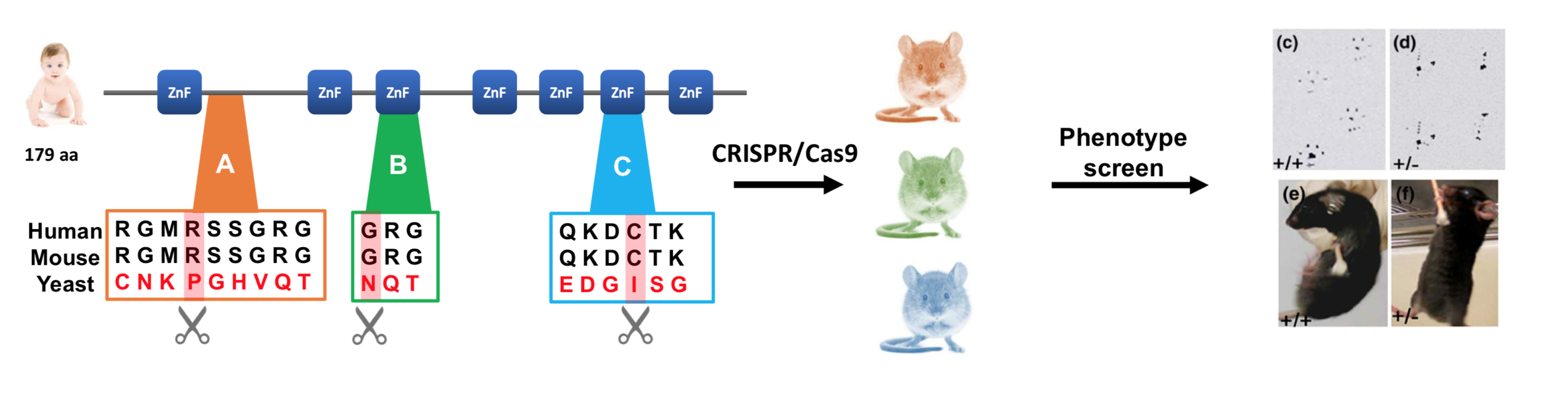
Figure 1. Workflow for identifying conserved sites of CNBP important to proper skeletal muscle function.
The sites that I will choose to mutate will be conserved sites that are only conserved within muscular organisms. Using the FASTA sequence for a non-muscular organism, like yeast, I will find regions within the yeast homolog that are not conserved when compared with other muscular species. I will then mutate the sites that are conserved from humans to mice, but not from mice to yeast, because I believe that these regions are the most likely to be implicated in muscle function. Since yeast does not have muscles, one would expect the regions important for muscle regulation not to be found in yeast. Therefore, I suspect that specific sites conserved only within muscular organisms will be critical in regulating muscle function.
Aim 2
The purpose of my second aim is to identify differentially expressed transcripts in the CNBP mutants that are important for skeletal muscle function. To do this, I will use RNA-seq on wild-type (WT) mice and the CNBP mouse mutants from Aim 1 to identify RNA transcript profiles in muscle tissue and determine any differentially expressed genes. Then, using Gene Ontology (GO), I will sort expressed genes by function and identify any differences in transcript levels between the normal and disease states. I will then knock out these genes using CRISPR to determine if they show the same muscle wasting and weakness phenotypes as the CNBP mutants
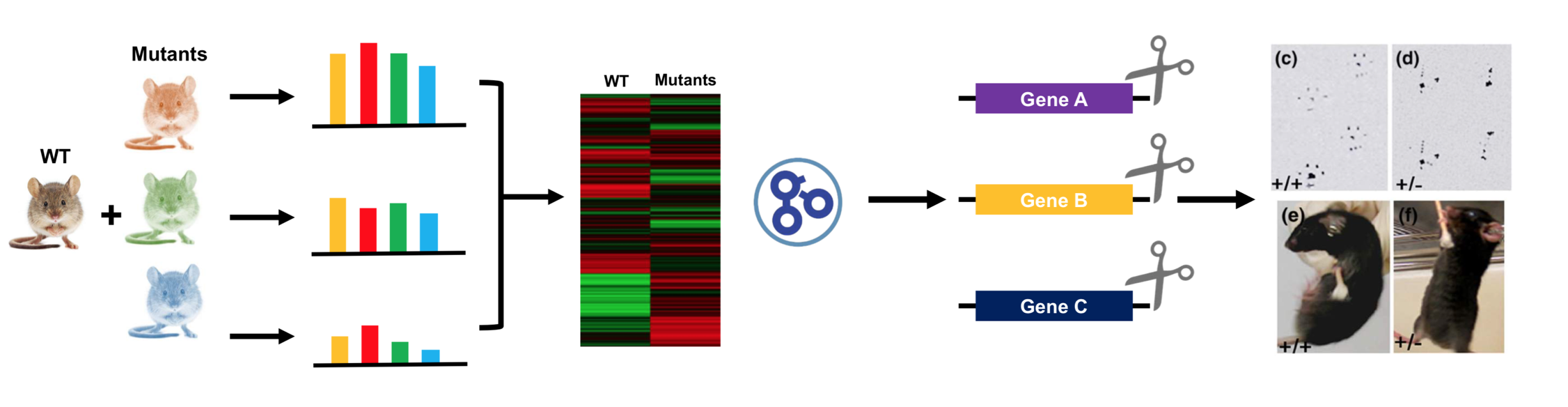
Figure 2. Workflow for identifying differentially expressed transcripts in CNBP mutants important for skeletal muscle function.
The overall purpose of conducting this screen will be to determine genes that may be transcriptionally regulated by CNBP. Since CNBP has been identified as a DNA binding protein, this screen would provide information about specific genes that CNBP may be regulating at the transcriptional level. This screen will also identify which genes specific to skeletal muscle function are up-regulated and down-regulated in the WT vs. mutant state.
Aim 3
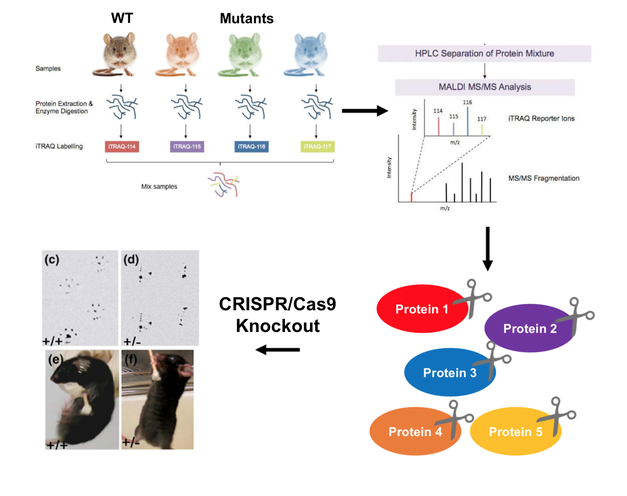 Figure 3. Workflow for quantifying the dysregulation of proteins involved in skeletal muscle function.
Figure 3. Workflow for quantifying the dysregulation of proteins involved in skeletal muscle function.
The purpose of my third and final aim is to quantify the dysregulation of proteins involved in skeletal muscle function. By using an iTRAQ method of proteomic analysis, I will isolate proteins found within the skeletal muscle of WT mice and the mice mutants from Aim 1 in order to quantify any up-regulated or down-regulated proteins in each state. Using PANTHER, I will classify these proteins by their GO terms to determine what protein classes and biological processes are affected in the disease state. Following this, I will then knock out any dysregulated proteins using CRISPR and screen for muscle weakness and wasting phenotypes.
This screen will determine any dysregulation in skeletal muscle genes at the post-transcriptional level caused by decreased CNBP levels. Since CNBP has been identified to also function as RNA binding protein, this screen would provide information about specific genes that CNBP may be regulating at the translational level. By identifying dysregulation at both the protein and mRNA level, I can see whether or not CNBP is acting as a transcriptional and translational regulator. This screen will also identify what protein classes are most affected via GO analysis. Ultimately, I suspect that CNBP mutants will have dysregulated proteins levels and that these proteins will be critical to skeletal muscle function.
Future Directions
For future directions, I believe it will be important to further characterize the role of CNBP and work identify its mRNA and protein targets. In addition to further studies of CNBP in muscle tissue, I think characterizing its interactions in other tissues, like eye and endocrine tissues, will be extremely important in being able to understand the wide range of symptoms the DM2 leads to.
Moving away from the CNBP protein itself, I think another important future direction would be to conduct chemical screens to remove toxic CNBP-pre-mRNA. This mutant pre-mRNA causes a whole host of detrimental effects, so a small molecule that could remove it would likely be a very beneficial drug.
Finally, I think continuing to develop new medications and therapies aimed at treating DM2 symptoms like muscle pain and myotonia will be very beneficial in improving the quality of life for people affected by DM2.
Moving away from the CNBP protein itself, I think another important future direction would be to conduct chemical screens to remove toxic CNBP-pre-mRNA. This mutant pre-mRNA causes a whole host of detrimental effects, so a small molecule that could remove it would likely be a very beneficial drug.
Finally, I think continuing to develop new medications and therapies aimed at treating DM2 symptoms like muscle pain and myotonia will be very beneficial in improving the quality of life for people affected by DM2.
Conclusion
Overall, DM2 is disease characterized by many symptoms. It is caused by excessive CCTG nucleotide repeats within the first intron of the CNBP gene. DM2 is the result of many molecular changes, but the effect that lower CNBP protein levels has on the disease remains unclear. Learning more about each of these mechanisms will ultimately help lead to treatments and hopefully an eventual cure for DM2 in the near future.
Final Presentation |
Final Presentation- First Draft |
|
| ||||
References
- Meola, G., & Cardani, R. (2015). Myotonic Dystrophy Type 2: An Update on Clinical Aspects, Genetic and Pathomolecular Mechanism. Journal of Neuromuscular Diseases,2(S2). doi:10.3233/jnd-150088
- CNBP CCHC-type zinc finger nucleic acid binding protein [Homo sapiens (human)] - Gene - NCBI. (n.d.). Retrieved from https://www.ncbi.nlm.nih.gov/gene/7555
